
Using genotype abundance to improve phylogenetic inference
I was amazingly fortunate to get to work my first project in phylogenetics under Erick Matsen (Fred Hutch) and Vladimir Minin (UW at the time, now UC Irvine). The aim was to incorporate genotype abundance information in phylogenetic inference for experimental systems in which unfolding evolutionary processes are sampled at single-cell resolution. For such data sets, we have counts of the number of individuals that bear each genotype—a situation that standard phylogenetics algorithms can’t handle (so this abundance information is generally ignored). Things began with chalkboard scribbles like this:
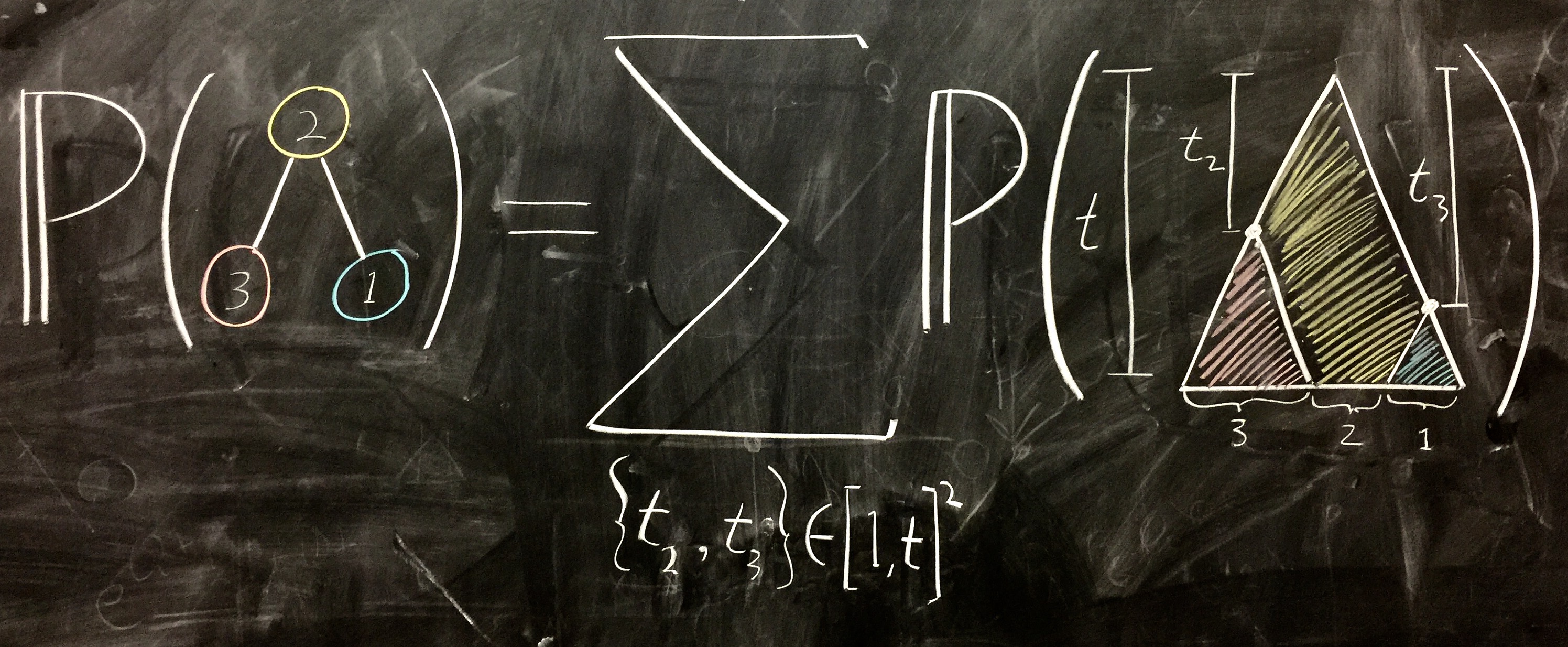
We used a mix of standard phylogenetics approaches and stochastic process likelihoods to capture the intuition that genotypes with higher observed abundance will tend to have more descendant genotypes. After validating with extensive simulations, I set out for The Rockefeller in NYC to collaborate with Luka Mesin and Gabriel Victora on empirical tests of our methods in the setting of B cell receptor affinity maturation. They have some amazing experimental windows into affinity maturation, just check out this video about Gabriel’s recent MacArthur “Genius” award:

Our validation efforts seem to show that we are productively integrating the abundance information to infer better trees. Although we were motivated by B cell affinty maturation, we’re hoping GCtree will be useful in other settings, such as single-cell tumor phylogenetics, and cell lineage tracing using genome editing (e.g. the GESTALT magic coming out of the nearby Shendure lab).
Here’s the repo for the GCtree software, and the arXiv preprint of our manuscript. We’d be very happy to hear from others interested in inferring phylogenetic trees for data with genotype abundance information, and I think we’ve done a decent job of making the code general and usable.
And check out Erick’s nice blog post about the project!